Next-generation precision antibody profiling; from science fiction to reality
The cost of drugs and targeted therapies is a major and growing challenge in the health system. Reducing the time taken to develop novel therapies will reduce costs to the health system.
To address this grand challenge, it is imperative to better understand how the human organism defends itself against diseases. The biggest mystery is the human immune system; understanding this ultimately requires knowledge of the amino acid sequence repertoire of human antibodies and their respective antigens.
Antibodies are the most sophisticated line of natural defence against disease. Knowing which antibodies are produced in response to a given disease enables us to understand the disease cause better and to provide next-generation cures in the form of personalised therapeutic antibodies. The limiting factor for this to truly be achieved is finding a way to sequence large molecules in the gas phase using mass spectrometry (MS), and this represents a formidable challenge.
Current state of the art
Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) is a key technique in the analysis of individual proteins as well as whole proteomes. Current state-of-the-art antibody analysis uses a bottom-up approach based on MS/MS sequencing of the peptides obtained after S-S bond reduction and enzymatic digestion of the antibody. Although it is now possible to obtain 100 per cent sequence coverage by bottom-up MS followed by intensive post-processing data analysis protocols, such analytical workflows can only be applied to single isolated antibodies. Extending current tools and methods to complex mixtures becomes impractical as reconstructing the different amino acid sequences using peptides originating from different antibodies present in the protein digest is practically impossible.
The solution is to keep proteins intact before MS analysis and perform sequencing in the gas phase; this is known as the top-down MS/MS approach. Complete sequencing of intact proteins with a molecular weight of more than 20 kilodalton (about 200 amino acids) still represents an insurmountable challenge to MS, mainly because of the restrictions imposed by MS/MS techniques as well as the inability to decipher all molecular information from mass spectra exhibiting massively overlapping isotopic distributions. Traditional collision-activated dissociation (CAD), higher-energy collisional dissociation (HCD) and electron transfer dissociation (ETD) employed in Orbitrap™™ and time-of-flight (TOF) mass spectrometers do not produce cleavage of all inter-residue bonds. Even UV photons employed in top-down MS/MS of antibodies cleave only 30–40 per cent inter-residue bonds at best, which is far from sufficient. Some attempts to cleave an antibody into a few larger (20 to 50 kilodalton) parts in a middle-down approach have shown promise but are still a long way from success in providing full sequence coverage.
The simple fact is that more energetic and diverse MS/MS fragmentation techniques are needed! The most promising are the non-ergodic reactions, where covalent bond breakage occurs faster than energy redistribution over the entire ion. An example of such a technique is electron-capture dissociation (ECD) (Zubarev Kelleher and McLafferty, 1998), in which multiply-charged protein polycations capture low-energy electrons. However, ECD releases only 4–7 eV of recombination energy, which is not enough. Another great challenge is data analysis. The most useful m/z range in Fourier transform (FT) mass spectra, as obtained from Orbitrap™ instrumentation, is from 200 to 2000, while the masses of antibody fragments are from 200 Da to 150 000 Da, and the charges range from 1+ to ≈75+. Because of the stable isotopes, such as 13C, 15N, etc., every molecule has an isotopic distribution, with the width varying from 2 Da to ≈25 Da. Every inter-residue link can be cleaved in at least three different ways, producing more than six terminal fragments—with many other possibilities as well. Thus the MS/MS spectrum is literally stuffed with hundreds of overlapping fragments ions, making it difficult to identify all, and even most, of them.
Beyond state-of-the-art: TopSpec
The TopSpec project aims to solve this challenge, opening up new opportunities in medical research and drug development that are only dreamt about today. The TopSpec project is developing a ground-breaking TOP-down tandem mass SPECtrometry (MS/MS) platform. In this project, key experts in radical-assisted sequencing of proteins have teamed up with the best developers in ultrahigh-resolution MS and MS/MS, as well as top European scientists in online protein separation and MS data processing. Achieving the seemingly impossible, we are creating, testing and validating a seamless platform capitalising on our ground-breaking innovations, enabling scientists from academia and industry to explore the antibody repertoire, enhancing our ability to explore new effective cures for major global diseases.
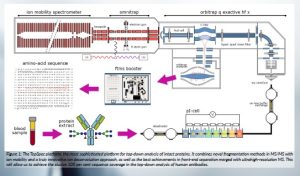
The targeted breakthrough of the TopSpec platform (Figure 1) is its unique ability to generate enhanced sequence information by analysing whole intact antibodies.
TopSpec objectives
- Sequence
The biggest problem in top-down MS/MS: sequencing of large proteins is addressed by implementing novel gas-phase radical reactions in the ground-breaking MS/MS device, the Omnitrap™ (Papanastasiou et al., 2022). An ion mobility (IM) device is attached to it, providing hardware deconvolution of overlapping isotopic clusters that present a daunting problem for deconvolution algorithms. The ions are then detected by an ultra-high resolution Orbitrap™ mass analyser with extended functionality. - Analyse
To solve the second biggest problem in top-down MS/MS: implementing novel deconvolution procedures to attribute isotopic peaks to individual molecules or fragments. The custom-designed FT Booster offers the most modern advances in signal acquisition and real-time data processing, increases the MS/MS spectra quality and radically simplifies them via our breakthrough deconvolution algorithm. - Optimise
To optimise the front-end, online separation of large proteins by replacing the conventional high-pressure reversed-phase liquid chromatography, poorly suited for large proteins, with a revolutionary novel low-pressure separation device called the pI-Trap. - Combine
To seamlessly combine the aforementioned components into a TopSpec instrumentation platform by designing sophisticated software to control the whole platform as a single apparatus. Novel software for data acquisition, processing and analysis, including de novo protein sequencing, is being created. - Utilise
Create novel top-down strategies that fully utilise the analytical power of TopSpec to sequence large proteins and implement these strategies for solving disease-related problems.
Antibodies are enriched from human blood at consortium partners Karolinska Institute (Stockholm, Sweden) and Institut Pasteur (Paris, France). They are loaded into the pI-Trap, an innovative micro-preparative device developed by Biomotif (Täby, Sweden). Upon desalting in an online desalinator, pI-Trap separates antibodies by their isoelectric point (pI) in solution. After online buffer exchange, they are ionised and transferred into the gas phase by electrospray ionisation (ESI). The desalination step is critical for obtaining good pI fractionation and obtaining a high abundance of molecular ions to enable MS/MS sequencing.
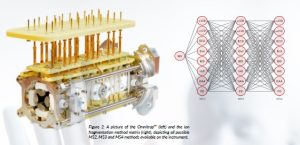
The antibody ions then enter a hybrid quadrupole Orbitrap™ MS where they are selected by m/z in a quadrupole filter of a Thermo Scientific™ Q-Exactive™ HF instrument (at Institut Pasteur) or Thermo Scientific™ Exploris™ 480 MS (at Karolinska Institute) and then sent onwards to the Omnitrap™. The Omnitrap™ (Figure 2), developed and built by Fasmatech (Athens, Greece), is a novel unique device, unprecedented in its flexibility and capabilities. Attached to the back end of the Orbitrap™ MS, it employs a hexapole ion guide followed by nine linear ion trap segments, with a full arsenal of ion manipulation abilities, such as m/z isolation, radial and translational ion motion excitation, and transfer between the traps in any order. The Omnitrap™ is equipped with an electron gun, a hydrogen atom source, as well as a UV light source, enabling all possible MS/MS fragmentation techniques, such as electron bombardment with attachment (ECD, ≈0 eV), excitation (1-10 eV) and ionisation (10-1000 eV energy range), hot hydrogen atom bombardment, plasma ion bombardment, UV photodissociation, and collisional dissociation both in the low- (CID) and higher-energy (HCD) ranges. Importantly, these MS/MS (MS2), MS/MS/MS (MS3), and MS/MS/MS/MS (MS4) approaches can be combined in any order, starting from HCD MS/MS that is already implemented in the Orbitrap™. The Omnitrap™ is able to perform multi-notch selection of several charge states of protein ions simultaneously, which progressively improves the analysis sensitivity, as large molecules appear in ESI in many charge states. After ion excitation and fragmentation, the remaining precursor ions can be selectively removed to reduce dynamic range problems and facilitate the detection of low-abundant fragments. The Omnitrap™ can also accumulate the MS/MS products from several MS/MS events for subsequent simultaneous detection with enhanced signal/noise, a critical design aspect for pushing the boundaries in top-down MS. The instrument control software allows the Omnitrap™ to operate seamlessly with the Orbitrap™ mass spectrometer.
To facilitate the analysis of the multitude of fragment ions in different charge states overlapping on the m/z scale, IM gas-phase fractionation will be implemented. IM uses the fact that the low-energy collision cross section of ions is larger for higher masses and charge states. Thus fragment ions from the Omnitrap™ will be ‘dragged’ through collision gas in the IM device and fractionated by their collisional cross sections into three fractions. Each fraction will be sent to the Orbitrap™ for detection and analysed individually.
To accommodate the extreme information flow, the Orbitrap™ ion detector is equipped with a novel version of the FTMS Booster, developed by Spectroswiss (Lausanne, Switzerland), including algorithms from Nottingham Trent University (Nottingham, UK), for increased sensitivity, resolution and mass accuracy. Critically, low-abundant signals are kept. FTMS Booster works in parallel with the conventional ion detection system of the Orbitrap™, ensuring system robustness. The device uniquely stores signal transients, allowing one to implement the novel deconvolution strategy based on the fact that the signal transient decays due to high-energy collisions with residual gas, and the decay rates for the individual ions of an isotopic cluster are similar, thus can be clustered using their decay constants.
In the next edition of PRj, we will present the exciting results we have obtained with the TopSpec platform.
References
Papanastasiou, D., Kounadis, D., Lekkas, A., Orfanopoulos, I., Mpozatzidis, A., Smyrnakis, A., Panagiotopoulos, E., Kosmopoulou, M., Reinhardt-Szyba, M., Fort, K., Makarov, A. and Zubarev, R. A. (2022) ‘The Omnitrap™ Platform: A Versatile Segmented Linear Ion Trap for Multidimensional Multiple-Stage Tandem Mass Spectrometry’, Journal of the American Society for Mass Spectrometry, 33(10), pp. 1990–2007. doi: 10.1021/jasms.2c00214.
Zubarev, R. A., Kelleher, N. L. and McLafferty, F. W. (1998) ‘Electron Capture Dissociation of Multiply Charged Protein Cations. A Non-ergodic Process’, Journal of the American Chemical Society, 120(13), pp. 3265–3266. doi: 10.1021/ja973478k.
Project name
TopSpec
Project summary
The purpose of the TopSpec project is to develop tools to establish the sequence repertoire of human antibodies and their respective antigens, to better understand how the human organism defends itself against diseases. Results obtained confirm that this is a breakthrough technology that will revolutionise academic, clinical and industrial proteomics and dramatically advance the development of antibody- and protein-based therapeutics.
Project partners
Karolinska Institute, Sweden; Fasmatech, Greece; Thermo Fisher Scientific, Germany; Spectroswiss, Switzerland; BioMotif, Sweden; Nottingham Trent University, United Kingdom; Institut Pasteur, France; Spectrometry Vision, The Netherlands.
Project lead profile
Roman Zubarev is a professor of medicinal proteomics in the Department of Medical Biochemistry and Biophysics at the Karolinska Institutet. His research focuses on the use of mass spectrometry in biology and medicine. In 1997, Zubarev discovered the phenomenon of electron-capture dissociation (ECD) of polypeptides. He later developed ECD and other ion-electron reactions as analytical techniques in Odense and Uppsala.
Project contacts
Prof. Dr Roman Zubarev
Biomedicum, 9A, floor 9, Solnavägen 9, 171 65 Solna, Sweden
+46 8 524 875 94
https://ki.se/en/mbb/roman-zubarev-group
Funding
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 829157.
Figures
Figure 1: The TopSpec platform, the most sophisticated platform for top-down analysis of intact proteins. It combines novel fragmentation methods in MS/MS with ion mobility and a truly innovative ion deconvolution approach, as well as the best achievements in front-end separation merged with ultrahigh-resolution MS. This will allow us to achieve the elusive 100 per cent sequence coverage in the top-down analysis of human antibodies.
Figure 2: A picture of the Omnitrap™ (left) and the ion fragmentation method matrix (right), depicting all possible MS2, MS3 and MS4 methods available on the instrument.